Polycystic Kidney Disease Causes and Treatment
| ✅ Paper Type: Free Essay | ✅ Subject: Chemistry |
| ✅ Wordcount: 3475 words | ✅ Published: 18 Aug 2017 |
Introduction:
Polycystic kidney disease effects roughly 10 million people worldwide. Even though this disease is so prominent it lacks research in the field of therapeutics from biopharmaceutical companies as they invest their resources into fields which are seen to be more profitable i.e. cancer research. This lack of research is what enticed us to carry out our project on polycystic kidney disease.
This project will firstly outline the history of the polycystic kidney disease, how it effects patients, the cause of the disease and the current therapeutic treatment available to combat polycystic kidney disease in section 1.
Section 2 will look at the current diagnostic methods employed by a physician to see if a patient is suffering from polycystic kidney disease. Diagnostic methods such as imaging and genetic testing will be dealt with here.
Lastly section 3 will look at a potential new diagnostic technique which has been formed using proteomic techniques to identify the difference between a healthy polycystin-1 protein compared to a mutated polycystin-1 protein.
The first record of Polycystic Kidney Disease (PKD) is from the 16th century. In 1586, the King of Poland died from cysts on his kidneys. The cysts were described by his surgeon as “large like those of a bull, with an uneven and bumpy surface”. At the time of his death he was diagnosed with meningeal abscesses. It wasn’t until a group of physicians re-examined the records of the King’s death over 300 hundred years later that his cause of death was agreed to be PKD. The term polycystic kidney disease was first used by Félix Lejars in 1888, although the mode of inheritance of this disease wasn’t understood for almost another one hundred years. In the 1990’s, the formation of cysts was understood at a molecular level. This helped in the discovery of the genes that cause PKD (Ayse, 2016).
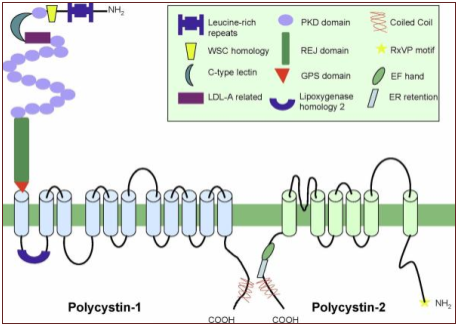
Figure.1 A visual representation of the transmembrane proteins Polycystin-1 and Polycystin-2. Also seen is the Polycystin-1 receptor located in extracellularly.
(Gallagher, Germino and Somlo, 2010)
PKD is a genetic disease in which the renal tissue within the outer cortex and inner medulla is replaced with fluid filled sacs (or cysts). These cysts enlarge the kidneys and inhibit kidney function. Hypertension, hematuria and chronic pain are the most common symptoms associated with PKD (Seeger-Nukpezah et al., 2015). PKD has two forms; Autosomal Dominant (ADPKD) and Autosomal Recessive (ARPKD). ADPKD is the result of the inheritance of one mutant PKD1 or PKD2 gene, which affects ~1:750 people worldwide. 85% of ADPKD cases are caused by mutations in PKD1. Mutations in this gene lead to earlier disease onset. The other 15% of cases are attributed to a mutation in PKD2.
All ADPKD patients inherit one normal allele and one mutant allele. Cases where both alleles have the dominant mutation lead to embryonic lethality. ARPKD is caused by the inheritance of two recessive mutated PKHD1 genes. ARPKD is considerably less common with an incidence of 1:20,000. ARPKD progresses at a much faster rate than ADPKD. It usually causes death at birth or requires transplantation in early childhood. Multiple different types of mutations in PKD1, PKD2, and PKHD1 have been researched, including frameshifts, deletions, and premature stop codon insertions (Wilson, 2015).
In the dominant form of PKD there is only one mutated gene inherited. This mutated gene is unable to produce the proteins PC-1 or PC-2. However, the non-mutated allele can still function as normal and can produce enough of the polycystin proteins to maintain kidney function. It is only when a somatic mutation occurs causing the normal allele to become inactivated that symptoms of the disease will occur. (Torres and Harris, 2010)
PKD-1 is located on the short arm of chromosome 16 (16p13.3). PKD-2 gene is located on the long arm of chromosome 4 (4q21) (Nowak et al., 2016). Polycystin-1 and 2 are large transmembrane proteins which are encoded by PKD1 and 2. Both proteins affect multiple downstream signalling proteins (Seeger-Nukpezah et al., 2015).
In a normally functioning nephron as the urinary filtrate flows by and causes the primary cilia to bend polycystin 1 and 2 respond by allowing calcium influx which activate pathways in the cell which inhibit cell proliferation. PC1 has the ability to sense when the primary cilia bends which activates PC2 calcium channels. If either PC1 or PC2 is absent the signal to inhibit cell growth is not received (Chebib et al., 2015).
PKD1 or PKD2 mutations cause a reduction in intracellular calcium. This triggers a change in the response of the cell to cyclic adenosine monophosphate (cAMP) from suppression to proliferation. The concentration of cAMP directly affects the activity of Protein Kinase A. Four cAMP molecules are required to activate one PKA enzyme. The increased production of cAMP via adenylyl cyclase 6 is dependent on vasopressin (Chebib et al., 2015).
Despite the many breakthroughs in research allowing for a greater understanding of the disease, there is currently no cure for PKD. However, there are drugs which can suppress symptoms brought on by the disease. Beta blockers such as, Tenormin, can be used to treat hypertension and haematuria can be treated with antibiotics. Understanding the effect of PC1 and PC2 mutations on the vasopressin receptor in the cell lead to the development of Tolvaptan. This drug slows down the formation of cysts in the kidneys. Tolvaptan blocks the vasopressin receptor, which will stop the signalling pathway. Therefore, cAMP production will be reduced. (Ema.europa.eu, 2017)
The current diagnostic methods for polycystic kidney disease include genetic testing, pre-natal testing and imaging studies in the form of ultrasounds, CT (Computed Tomography) scans and MRI (Magnetic Resonance Imaging) scans
The imaging studies take a scan of the kidneys to identify the presence of any abnormalities in the form of renal cysts. An ultrasound mechanism uses high frequency sound waves to capture and visualise images that can’t be seen with the naked eye. The CT scan combines many x-ray images with the aid of a computer to generate cross sectional views and/or three-dimensional images of the kidneys and the MRI scan uses a magnetic field and pulses of radio wave energy to from pictures of the kidneys.

The appearance of three or more renal cysts, either unilateral or bilateral, on the image is enough to diagnose a patient between the ages of 15 and 39 with polycystic kidney disease. In patients aged 40 – 59, the presence of two or more cysts in each kidney fulfils the criteria to diagnose the patient with polycystic kidney disease. The presence of four or more cysts in each kidney is used to diagnose older patients (F. Belibi et al., 2008). The kidneys on an image may appear enlarged but retain their normal reniform shape in the case of a patient presenting with possible polycystic kidney disease. The medullary pyramids in the centre of the kidney may be more visible on an image in contrast to the cortex which can give a peripheral halo on the image obtained. High resolution imaging studies allows the visualisation of numerous cylindrical cysts within the medulla and the cortex which represent ectatic collecting ducts within the kidney (F. Gaillard, 2015).
Genetic testing for polycystic kidney disease is for those who have a family history of polycystic kidney disease who has no symptoms and may consider being screened for the disease. Genetic tests can be done to screen for both PKD1 or PKD2 mutations. A method of PCR known as PCR- SSCP (Polymerase Chain Reaction – Single Strand Conformation Polymorphism) is used to view mutations, if any, in the patient’s genomic DNA. In SSCP analysis, a mutated DNA sequence is detected as a change of mobility in polyacrylamide gel electrophoresis caused by the altered folded structure of single-stranded DNA (K. Hayashi, 1991). The genomic DNA of the white blood cells in patients with the possible polycystic kidney disease gene are isolated. These samples of genomic DNA are then amplified by PCR using two primers to amplify the potential polycystic kidney disease genes (R. Jas et al., 2012) The PCR product is then analysed using the SSCP method. This method involves loading the PCR product samples onto the acryl amide gel and gel electrophoresis occurs. After completion of the gel electrophoresis step, the gel is subjected to silver staining to visualise the SSCP band patterns (B. Yadav et al., 2009). The silver stained gel is kept on a transilluminator and the SSCP variants are recorded. DNA samples from the abnormal bands seen on the transilluminator are sequenced to see what kind of mutation and where the location of the mutation is on the polycystic kidney disease gene (Z. Dian-Yong et al., 2002). In PCR-SSCP analysis, changes in several hundred base pairs are detected in contrast with other techniques in which changes in relatively short sequences can be detected. Because of this, PCR-SSCP analysis is much more sensitive to the replication errors that can occur during the PCR process (K. Hayashi, 1991).
Diagnosis of polycystic kidney disease can also be done prenatally. If the parents agree, a prenatal diagnosis can be done on the developing fetus if there is a history of polycystic kidney disease in either the parents or extended family . A DNA sample is taken from both parents and a sample of tissue is taken from the fetus. The tissue sample is obtained from the fetus by a procedure called aminocentesis which involves passing a needle into the mother’s lower abdomen and into the amniotic cavity inside the uterus. The sample is then amplified by PCR to detect any mutations in the DNA that could lead to the fetus developing polycystic kidney disease in the future (K. MacDermot et al., 1998).
The imaging studies, genetic testing and prenatal testing for polycystic kidney disease have advantages and disadvantages. One advantage of the imaging studies is that they are reliable, inexpensive and a non-invasive way to diagnose polycystic kidney disease (A.Khan, 2015). A disadvantage of imaging studies is that while they are sensitive in the detection of polycystic kidney disease, problems may arise with smaller cysts. Smaller cysts on scans may not be easily differentiated from small, solid masses within the kidneys (A.Khan, 2015). An advantage of CT scans when compared to MRI scans is that the cysts on the kidney will enhance on the image when dye is administered into the patient intravenously (A. Khan, 2015) (F. Gaillard, 2015). MRI scans of the kidneys are becoming a useful technique in diagnosing more complicated cysts and can be used in addition to or instead of CT scans (A. Khan, 2015).
The advantage of genetic testing as a method of diagnosing polycystic kidney disease is that it can determine if a person who has a relative with polycystic kidney disease will in the future start showing symptoms of the disease. Some disadvantages of genetic testing as a method of diagnosing polycystic kidney disease is that they are extremely costly tests to carry out and sometimes they can’t pick up on certain gene mutations that could eventually lead to the person having polycystic kidney disease (National Kidney Foundation, 2016).
The major advantage of prenatal testing for polycystic kidney disease is the fact that treatment using cyst suppressing drugs can be used early in the diseased patient’s life meaning the formation of renal damaging cysts will be slowed down drastically compared to a patient who was not on the cyst supressing treatment early in their life time. The negative of prenatal testing is of course the invasive nature of extracting tissue from the foetus which many parents would not agree with however the benefits of early diagnosis of this disease will lead to a better quality of life for the child in their later years.
As discussed in the previous section the current diagnostic methods for diagnosing polycystic kidney disease is through the use of various scans and genetic testing. The genetic testing is carried out by analysing the DNA sequence in order to identify any mutations which may be present.
As a new method of diagnosing this disease a study of the protein polycystin-1 which when mutated is responsible for polycystic kidney disease, will be analysed by using proteomic methods.
Firstly, the polycystin-1 protein must be extracted from a patient who wishes to obtain diagnosis of the disease. Since polycystin-1 is a membrane protein and is located in the kidney it will be necessary to extract kidney tissue from the patient by carrying out a quick and simple biopsy procedure. The biopsy removes kidney tissue by inserting a thin biopsy needle through the skin and into the kidney whilst the patient is under local anaesthetic.
Now that tissue containing the polycystin-1 or its mutated form is extracted from the patient it must be treated in order to release the proteins contained within the tissue. The tissue will firstly be homogenized and lysed in order to release the proteins into solution. The sample will then be centrifuged at 14,000 rpm at 4ÌŠC for 15 min. This centrifugation step removes any insoluble material and the supernatant will contain the proteins from the tissue sample including the protein of interest polycystin-1. This method was carried out by Malhas, Abuknesha, and Price 2001 whilst trying to crystalize the polycystin-1 protein.
Now that polycystin-1 is in solution it can be separated from the other proteins by means of 2D gel electrophoresis. This technique will separate the polycystin-1 protein from other proteins based firstly on their isoelectric points (pIs) and secondly by their molecular weight. After carrying out this two-dimensional separation the gel is stained with stains such as coomassie brilliant blue (CBB) or silver staining in order to visualize the spots on the gel. By carrying out bioinformatical analysis the molecular weight and pI of polycystin-1 can easily be obtained (Mishra 2010).
The molecular weight of polycystin-1 is 460.3 kDa and it has a pI of 6.27. With this information, the band which corresponds to the molecular weight and pI of polycystin-1 can be easily identified and excised from the gel.
Before excising the polycystin-1 protein from the gel it must first be fragmented into peptides using trypsin cleavage.
By fragmenting the protein with trypsin, peptide molecules are formed which are now suitable to be sequenced using MALDI-TOF mass spectrometry. These peptides must then be suspended in a matrix suitable for MALDI-TOF MS. An example of such a matrix is α-cyano-4-hydroxycinnamic acid which was used in the work carried out by Malhas, Abuknesha, and Price, when they crystallized the polycystin-1 protein using the MALDI-TOF technique. This matrix is suitable as the peptides are below 5 kDa.
The matrix and peptide mixture is then loaded onto the metal plate of the MALDI-TOF MS analyser where it will be hit by a pulsing UV laser. The matrix molecules absorb the UV light causing the matrix molecules to enter the gas phase along with their coupled vaporised peptides which then become ionized. The TOF MS then measures the time it takes the ions to fly as lighter ions travel faster. The ions will then hit the ion detector and the on board computer will produce the plot of the mass spectrum (Kraj and Silberring 2008).
MALDI-TOF MS is the ideal method to analyse the polycystin-1 protein as like ESI, it is a soft ionization technique meaning there will be little or no fragmentation of the compounds being analysed. Once the mass spectrum is obtained it is then compared to the mass spectrum of the normally functioning polycystin-1 protein which can theoretically be fragmented by trypsin in the online databases. Any differences between the sample and the known mass spectrum of the normal protein will signal a mutation has occurred.
This method of protein analysis is a very effective and efficient way of screening for the mutated proteins. The study carried out by Brioude et al 2016 proves this as they looked at using MALDI-TOF MS to test for mutated proteins leading to lung tumours. In this study, they concluded that the method is very promising and it should be used in several surgical settings where rapid evaluation of abnormal tissue is required. This highlights how this new method of analysing polycystin-1 for a mutation could prove very effective in diagnosing polycystic kidney disease.
This method holds a distinct advantage over the imaging methods currently in use as diagnosis can be made early in a patient’s life before they show symptoms or the formation of cyst on their kidneys which are associated with polycystic kidney disease. Early diagnosis of this disease means that drugs for the prevention of cyst formation such as Jinarc© which contains the API Tolvaptan can prolong a patient’s kidney function by slowing down the rate at which the fluid filled cysts on the kidneys are formed.
This method is used to identify a mutation of the polycystin-1 protein which attributes to 85% of autosomal dominant polycystic kidney disease cases. The other 15% is made up of mutations of the protein polycystin-2. Fibrocystin is the mutated protein responsible for autosomal recessive polycystic kidney disease. Both the dominant and recessive forms of this disease can be diagnosed using the above method in order to identify a mutation in either polycystin-2 or fibrocystin proteins if there is no mutation of polycystin-1 observed.
From the above sections, it can be seen how sever and prominent polycystic kidney disease is worldwide. Although this disease is genetically inherited from or both parents, symptoms of the disease are slow progressing. In 85% of cases the disease will not advance to renal failure until the patient is 50-60 years of age.
The current diagnostic techniques currently used by doctors such as imaging and genetic testing have their benefits however they are also flawed. The major disadvantage seen in this project regarding the current diagnostic techniques is the diagnosis of polycystic kidney disease in a patient where uncontrolled cyst formation has already occurred and serious renal problems have begun.
With the use of our new proteomic technique earlier diagnosis will be possible before the disease has progressed to renal failure. Early diagnosis means that the use of drugs such as Tolvaptan© can be used to significantly slow down cyst formation which will ultimately increase the length of time a patient suffering with polycystic kidney disease has before renal failure occurs.
A.Khan (2015) ‘Imaging in Autosomal Dominant Polycystic Kidney Disease’, Medscape Journal , 01(1).
Ayse, B. (2016). Tear drops of kidney: a historical overview of Polycystic Kidney Disease. Giornale Italiano di Nefrologia, 1.
B. Yadav & D. Kale (2009) ‘Single Strand Conformation Polymorphism (SSCP) Analysis by Nondenaturing PAGE’ Journal of Biological Methods, 01 (1).
Brioude, G., Brégeon, F., Trousse, D., Flaudrops, C., Secq, V., De Dominicis, F., Chabrières, E., D’journo, X.-B., Raoult, D. and Thomas, P.-A. (2016) ‘Rapid diagnosis of lung tumors, a Feasability study using Maldi-Tof mass Spectrometry’, PLOS ONE, 11(5). doi: 10.1371/journal.pone.0155449.
Chebib, F., Sussman, C., Wang, X., Harris, P. and Torres, V. (2015). Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nature Reviews Nephrology, 11(8), pp.451-464.
Ema.europa.eu. (2017). European Medicines Agency – Human medicines – EU/3/13/1175. [online] Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/orphans/2013/09/human_orphan_001257.jsp&mid=WC0b01ac058001d12b&source=homeMedSearch [Accessed 4 Mar. 2017].
F. Gaillard (2015) ‘Autosomal Recessive Polycystic Kidney Disease’ Radiopaedia Journal, 01(1).
F.Belibi & C. Edelstein (2008) ‘Unified Ultrasonic Diagnostic Criteria for Polycystic Kidney Disease’ Journal of the American Society of Nephrology, 20 (1).
Gallagher, A., Germino, G. and Somlo, S. (2010). Molecular Advances in Autosomal Dominant Polycystic Kidney Disease. Advances in Chronic Kidney Disease, 17(2), pp.118-130.
K.Hayashi (1991) ‘PCR-SSCP: A Simple and Sensitive Method for Detection of Mutations in the Genomic DNA’ Genome Research, 01 (1).
Kraj, A. and Silberring, J. (eds.) (2008) Proteomics: Introduction to methods and applications. Chichester, United Kingdom: John Wiley & Sons.
MacDermot, K., Saggar-Malik, A., Economides, D. and Jeffery, S. (1998). Prenatal diagnosis of autosomal dominant polycystic kidney disease (PKD1) presenting in utero and prognosis for very early onset disease. Journal of Medical Genetics, 35(1), pp.13-16.
Malhas, A.N., Abuknesha, R.A. and Price, R.G. (2001) ‘Polycystin-1: Immunoaffinity isolation and characterisation by mass spectrometry’, FEBS Letters, 505(2), pp. 313-316. doi: 10.1016/s0014-5793(01)02842-3.
Mishra, N.C. (2010) Introduction to proteomics: Principles and applications. United Kingdom: John Wiley & Sons.
National Kidney Foundation (2016) Polycystic Kidney Disease [online], available: https://www.kidney.org/atoz/content/polycystic/ [accessed 26 February 2017].
Nowak, M., Huras, H., Wiecheć, M., Jach, R., RadoÅ„-Pokracka, M. and Górecka, J. (2016). Autosomal dominant polycystic kidney disease diagnosed in utero. Review. Ginekologia Polska, 87(8), pp.605-608.
Rusni Mohd, Jas (2012) ‘Amplification of Real Time High Resolution Melting Analysis PCR Method for Polycystic Kidney Disease (PKD) Gene Mutations in Autosomal Dominant Polycystic Kidney Disease Patients’ African Journal of Biotechnology, 11(25).
Seeger-Nukpezah, T., Geynisman, D., Nikonova, A., Benzing, T. and Golemis, E. (2015). The hallmarks of cancer: relevance to the pathogenesis of polycystic kidney disease. Nature Reviews Nephrology, 11(9), pp.515-534.
Torres, V. and Harris, P. (2010). Comprehensive Clinical Nephrology. 4th ed. Elsevier Saunders, pp.529-542.
Wilson, P. (2015). Therapeutic targets for polycystic kidney disease. Expert Opinion on Therapeutic Targets, 20(1), pp.35-45.
Z. Dian-Yong, Z. Shu-Zhong, T. Bing, Z. Wei-Li, D. Bing, S. Mao, S. Tian-Mei & M. Chang-Lin (2002) ‘Detection of Polycystic Kidney Disease Gene 2 Mutations in the Hans by PCR-SSCP’ Academic Journal of Second Military Medical University, 01 (1).
Cite This Work
To export a reference to this article please select a referencing stye below:
Related Services
View all


DMCA / Removal Request
If you are the original writer of this essay and no longer wish to have your work published on UKEssays.com then please click the following link to email our support team:
Request essay removal